DCTD: BIOMETRIC RESEARCH PROGRAM |
|
Development of bioinformatic methods for vaccine design
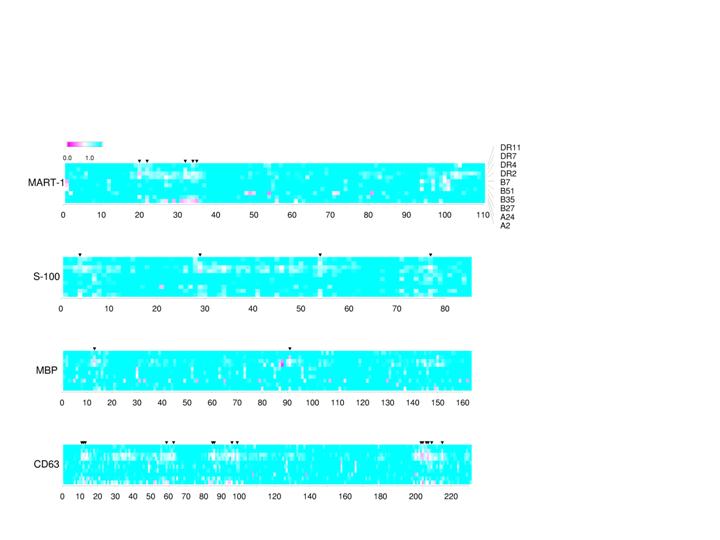
To design effective and specific vaccines, it is critical to identify the parts of the target (tumor or pathogen) that elicit strong immune responses (epitopes). We have developed a novel method to predict such epitopes (major histocompatibility complex protein binding peptides) that activate T lymphocytes, a major immune component. The computational models of MHC binding peptides can help facilitate the resource-consuming effort of antigenic peptide or epitope identification. Such bioinformatic approaches allow in silico scanning transcriptomes of tumor cells or proteomes of pathogens for putative T cell epitopes. For example, sometimes a tumor antigen arises due to its abnormal over-expression: mutated P53 proteins become stable and accumulate in many tumors. Different versions of P53 can be analyzed to reveal how their epitopes are affected by the mutations. We also use visualization of the putative epitope profiles to assess polymorphism effects of host and/or antigen (see example below).
|
|
Please send comments and suggestions to |
Updated on Nov. 2, 2015 |